
Doença genética rara e mortal tratada com sucesso no útero pela primeira vez
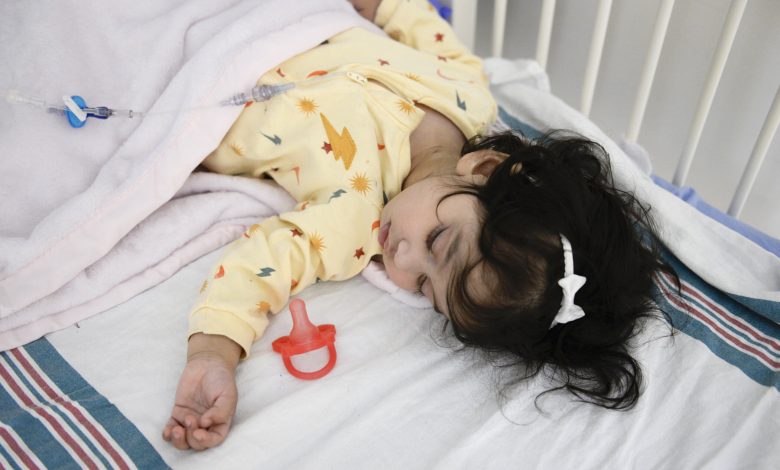

Paciente Ayla Bashir. Crédito: Universidade da Califórnia, São Francisco
Usando um protocolo desenvolvido na UC San Francisco, os médicos trataram com sucesso um feto com um distúrbio genético devastador pela primeira vez, e a criança agora está prosperando como uma criança, um estudo de caso no Jornal de Medicina da Nova Inglaterra relatórios.
“Este tratamento expande o repertório de terapias fetais em uma nova direção”, disse o co-autor sênior e correspondente Tippi MacKenzie, MD, cirurgião pediátrico do Hospital Infantil UCSF Benioff, codiretor do Centro de Medicina de Precisão Materno-Fetal da UCSF e diretor do Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research. “À medida que novos tratamentos se tornam disponíveis para crianças com condições genéticas, estamos desenvolvendo protocolos para aplicá-los antes do nascimento”.
O distúrbio da criança, a doença de Pompe de início infantil, é uma das várias doenças de armazenamento lisossomal que começam a causar danos graves aos principais órgãos, como o coração, antes do nascimento. Ao iniciar a terapia de reposição enzimática durante desenvolvimento fetalos médicos buscavam melhores resultados do que os típicos do tratamento pós-parto – resultados que podem incluir morte na primeira infância, tônus muscular muito baixo ou dependência de ventilador.
Após seis tratamentos pré-natais de reposição enzimática no Hospital de Ottawa, a criança, Ayla, nasceu a termo. Ela está recebendo terapia enzimática pós-natal no CHEO (um hospital pediátrico e centro de pesquisa em Ottawa, Canadá), e está indo bem aos 16 meses de idade. Ela tem função cardíaca e motora normal e está cumprindo os marcos de desenvolvimento.
“Quando tivemos Ayla, não sabíamos se ela conseguiria andar”, disse Zahid Bashir, pai de Ayla. “Não sabíamos se ela seria capaz de falar. Não sabíamos se ela seria capaz de comer. Não sabíamos se ela seria capaz de rir. Esses marcos, continuamos a nos surpreender com o progresso dela. Então, sim, é algo bastante, acho que às vezes podemos dar como certo, mas na maioria das vezes estamos cientes de que ela é um milagre.”
Um triunfo da colaboração
O sucesso do tratamento é um feito de colaboração entre a UCSF, onde se baseia um ensaio clínico em andamento sobre o tratamento; CHEO e The Ottawa Hospital, onde o paciente foi diagnosticado e tratado; e a Duke University, sede dos maiores especialistas do mundo em doença de Pompe.
“Nós realmente precisávamos deste grupo multidisciplinar de pessoas para emprestar sua profunda experiência em todos os aspectos do atendimento”, disse MacKenzie, que detém uma cátedra Benioff UCSF em Saúde Infantil e uma cátedra John G. Bowes em Biologia de Células-Tronco e Tecidos. “A terapia de reposição enzimática é uma nova fronteira no campo da terapia fetal; foi emocionante vê-la crescer a partir de um projeto de pesquisa em meu laboratório para impactar o resultado desta família em última análise. A UCSF é considerada o berço da cirurgia fetal e é um privilégio especial para nós continuar a expandir as tecnologias e tratamentos disponíveis para ajudar as famílias que enfrentam um diagnóstico difícil durante a gravidez.”
Em circunstâncias normais, a família do paciente teria viajado para o Centro de Tratamento Fetal do Hospital Infantil de Benioff da UCSF para participar do ensaio clínico. Quando as restrições do COVID-19 inviabilizaram as viagens internacionais, especialistas dos dois hospitais canadenses e dois americanos reuniram-se com a família por vídeo para discutir alternativas. A UCSF compartilhou o protocolo de tratamento com a equipe em Ottawa, onde a família mora. Durante todo o processo, toda a equipe se reuniu semanalmente por vídeo para discutir a saúde da mãe e do feto e acompanhar a resposta ao tratamento.
“Tratamos nossos pacientes fetais usando terapia intrauterina há mais de 30 anos”, disse Karen Fung-Kee-Fung, MD, especialista em medicina materno-fetal da família no Hospital de Ottawa e professora de obstetrícia e ginecologia da Universidade de Ottawa. . “O surgimento de um novo tratamento médico para aliviar o fardo da doença de Pompe para esta família e potencialmente ajudar outras famílias afetadas por doenças genéticas devastadoras é emocionante e incrivelmente satisfatório. colaboração para ajudar a tornar este tratamento inédito no mundo uma realidade.”
Pranesh Chakraborty, MD, pediatra e geneticista metabólico do CHEO e co-líder do estudo de caso, presta cuidados à família há anos. “Este tratamento é muito promissor e estou muito feliz por Ayla e sua família”, disse Chakraborty, que também é pesquisador do Instituto de Pesquisa CHEO e professor associado da Universidade de Ottawa. “Tendo tido o privilégio e o desgosto de caminhar ao lado de famílias que perderam filhos para essas doenças devastadoras, este trabalho é muito importante para mim”.
Um grande passo em frente para a terapia fetal
Os bebês nascidos com a doença de Pompe de início infantil geralmente têm corações aumentados e morrem dentro de dois anos se não forem tratados. A doença é muito rara, vista em menos de 1/100.000 nascidos vivos, e é causada por mutações em um gene que produz a alfa-glicosidase ácida, uma enzima que degrada o glicogênio. Sem ele ou com quantidades limitadas, o glicogênio se acumula perigosamente no corpo.
“Pelo nosso trabalho de longa data na Duke tratando pacientes com a doença de Pompe, sabemos em primeira mão a importância crítica do início precoce do tratamento”, disse Jennifer Cohen, MD, co-autora principal do estudo e professora assistente na Divisão de Genética Médica no Departamento de Pediatria da Duke University School of Medicine. “Nossa capacidade de oferecer uma nova oportunidade de tratamento a esta família e potencialmente mudar o curso desta doença difícil tornou esta colaboração e projeto inovador”, acrescentou Cohen.
Duke desempenhou um papel fundamental em muitos avanços no campo da doença de Pompe, incluindo o desenvolvimento de alglucosidase alfa como a primeira terapia de reposição enzimática (ERT) aprovada pela Food and Drug Administration (ERT) para a doença de Pompe, identificando o papel de títulos de anticorpos altos e sustentados para o ERT, usando biomarcadores para acompanhar a resposta ao tratamento e estabelecendo protocolos de indução de tolerância imunológica para os pacientes mais graves, observou Priya Kishnani, MD, coautora sênior, chefe da divisão de Genética Médica da Duke University School of Medicine. “Todos esses avanços foram cruciais para o tratamento e a resposta desse paciente em particular”, disse Kishnani. “A terapia intrauterina representa uma nova fronteira para pacientes com doença de Pompe.”
Pompe é uma das oito doenças de armazenamento lisossomal que a UCSF recebeu aprovação da FDA para tratar com terapia de reposição enzimática no útero para um ensaio clínico de Fase 1 de 10 pacientes. As outras doenças são Mucopolissacaridose tipos 1, 2, 4a, 6 e 7, doença de Gaucher tipos 2 e 3 e doença de Wolman.
Os pesquisadores esperam que o sucesso desta primeira aplicação e publicação do estudo de caso aumente a conscientização sobre o ensaio clínico da UCSF entre os pais com risco conhecido de transmitir essas doenças e os médicos que as tratam.
“Vendo o quão bem Ayla está, é importante buscar testes clínicos para estabelecer se esse tipo de terapia fetal será uma boa opção para outras famílias quando o tratamento no período neonatal não for suficientemente precoce”, disse Chakraborty, que dirige o programa provincial Newborn Screening Ontario, baseado no CHEO. “Estamos trabalhando duro para tentar melhorar o acesso a este teste para outras famílias canadenses”.
Dois pacientes adicionais com diferentes doenças lisossômicas foram agora inscritos no ensaio clínico da UCSF e ambos completaram seu curso pré-natal terapia de reposição enzimática. A primeira paciente deu à luz no final de outubro de 2022 e a segunda dará à luz no início de novembro de 2022. Ambas estão bem.
“É emocionante continuar esta pesquisa, que é um passo importante na evolução da terapia fetal, desde a cirurgia para condições anatômicas até terapias médicas para condições genéticas”, disse MacKenzie.
Jennifer L. Cohen et al, Terapia de reposição enzimática no útero para a doença de Pompe de início infantil, Jornal de Medicina da Nova Inglaterra (2022). 10.1056/NEJMoa2200587. www.nejm.org/doi/full/10.1056/NEJMoa2200587
Fornecido por
Universidade da Califórnia, São Francisco
Citação: Doença genética rara e mortal tratada com sucesso no útero pela primeira vez (2022, 10 de novembro) recuperada em 10 de novembro de 2022 em https://medicalxpress.com/news/2022-11-rare-deadly-genetic-disease-successfully.html
Este documento está sujeito a direitos autorais. Além de qualquer negociação justa para fins de estudo ou pesquisa particular, nenhuma parte pode ser reproduzida sem a permissão por escrito. O conteúdo é fornecido apenas para fins informativos.

