
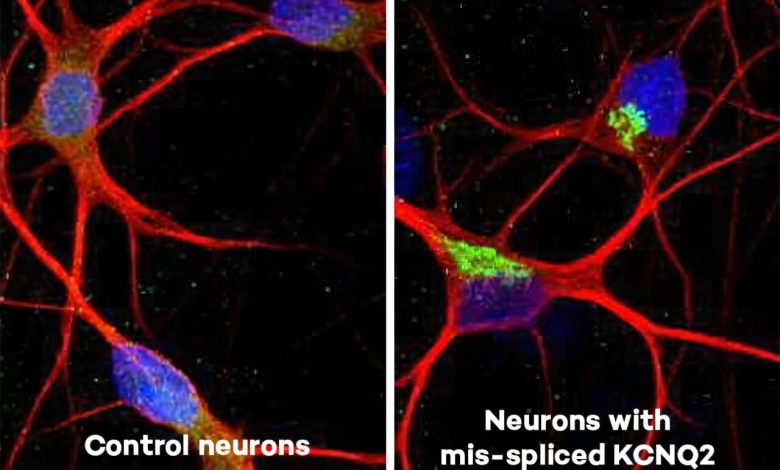
 em comparação com os neurônios do estudo com o gene KCNQ2 mal emendado. Crédito: Universidade Northwestern")
A imagem lado a lado mostra os neurônios de controle do estudo (à esquerda) em comparação com os neurônios do estudo com o gene KCNQ2 mal emendado. Crédito: Universidade Northwestern
Um novo estudo da Northwestern University usando tecido nervoso de pacientes e neurônios humanos cultivados em laboratório descobriu como uma proteína-chave da doença, TDP-43, impulsiona células nervosas hiperativas nas doenças neurodegenerativas esclerose lateral amiotrófica (ELA) e demência frontotemporal (DFT).
As descobertas não apenas explicam um mistério de longa data sobre o motivo pelo qual as células nervosas disparam na ELA e na DFT, mas também destacam um novo medicamento promissor para retardar ou prevenir a progressão da doença.
As descobertas são publicadas em Neurociência da Natureza em um artigo intitulado “A emenda incorreta de KCNQ2 dependente de TDP-43 desencadeia hiperexcitabilidade neuronal intrínseca em ELA/FTD”.
A ELA, que ataca os neurônios motores da medula espinhal e causa fraqueza progressiva e atrofia muscular, afeta cerca de 350 mil pessoas em todo o mundo. A DFT leva à atrofia dos lobos frontal e temporal do cérebro, que controlam a personalidade, o comportamento e a linguagem. Estima-se que entre 1,2 e 1,8 milhões de pessoas em todo o mundo vivam com alguma forma de DFT.
Embora a ELA e a DFT sejam bastante diferentes, uma característica consistente, mas mal explicada, de ambas as doenças é a hiperexcitabilidade neuronal, na qual os neurónios disparam demasiado e com demasiada facilidade.
Pesquisas anteriores descobriram que quase todos os casos de ELA e até metade dos casos de DFT partilham um problema característico: a proteína TDP-43 move-se da sua localização normal, o núcleo, para o citoplasma, e perturba a função celular normal.
O novo estudo descobriu que quando o TDP-43 funciona mal, ele interrompe a emenda normal do canal KCNQ2, que pode ser considerado um “freio” que normalmente impede que os neurônios disparem demais. Sem esse freio, os neurônios tornam-se eletricamente hiperativos (hiperexcitabilidade neuronal). Foi demonstrado que pacientes com ELA que apresentam hiperexcitabilidade apresentam risco aumentado de mortalidade, ressaltando seu significado clínico.
Os autores do estudo projetaram, desenvolveram e testaram um medicamento direcionado ao gene que, segundo o estudo, pode corrigir esse erro em neurônios humanos cultivados em laboratório, o que restaura o equilíbrio e reduz a hiperatividade.
O medicamento é conhecido como oligonucleotídeo antisense (ASO), que, uma vez clinicamente validado e aprovado, seria administrado por injeção direta no sistema nervoso central do paciente, semelhante a uma epidural.
“Ao corrigir o erro de splicing do KCNQ2 com o medicamento ASO, fomos capazes de acalmar neurônios hiperativos, e restaurar a atividade neuronal poderia potencialmente retardar a progressão da doença”, disse o autor correspondente Evangelos Kiskinis, professor associado de neurociência e neurologia na divisão de doenças neuromusculares na Northwestern University Feinberg School of Medicine.
“Estou emocionado por finalmente termos resolvido um mistério de longa data sobre por que as células nervosas na ELA/DFT estão hiperativas e estressadas antes mesmo de morrerem”.
Os cientistas estudaram neurônios de pacientes e cultivados em laboratório, bem como ELA pós-morte e tecido cerebral e da medula espinhal FTD. O defeito que descobriram é específico dos seres humanos e não ocorre em modelos de camundongos ou ratos, disse Kiskinis.
Os pacientes no estudo com splicing incorreto de KCNQ2 mais grave tiveram início mais precoce da doença, descobriu o estudo, tornando-o um possível biomarcador para prognóstico ou resposta ao tratamento, disse Kiskinis.
“Nosso trabalho conecta duas características centrais da doença – patologia TDP-43 e hiperexcitabilidade – em um único caminho mecanicista”, disse Kiskinis. “Isso também aponta para um novo e excitante alvo terapêutico.”
A equipe de Kiskinis está atualmente trabalhando para desenvolver um teste de biomarcador baseado na identificação desse evento KCNQ2 mal processado que poderia levar a um diagnóstico mais precoce.
“Estamos entusiasmados em levar este ASO para estágios clínicos”, disse ele.
Outros autores do estudo da Northwestern incluem Kelly A. Marshall, estudante de pós-graduação em neurociência interdepartamental da Northwestern University, Francesco Alessandrini, pós-doutorado, e Dr. Alfred L. George, presidente do departamento de farmacologia.
Mais informações:
O splicing incorreto de KCNQ2 dependente de TDP-43 desencadeia hiperexcitabilidade neuronal intrínseca em ELA/FTD, Neurociência da Natureza (2025). DOI: 10.1038/s41593-025-02096-w.
Fornecido pela Universidade Northwestern
Citação: Nova pista para ALS e FTD: proteína defeituosa interrompe o sistema de ‘freio’ do cérebro (2025, 31 de outubro) recuperada em 31 de outubro de 2025 em https://medicalxpress.com/news/2025-10-clue-als-ftd-faulty-protein.html
Este documento está sujeito a direitos autorais. Além de qualquer negociação justa para fins de estudo ou pesquisa privada, nenhuma parte pode ser reproduzida sem permissão por escrito. O conteúdo é fornecido apenas para fins informativos.
